Prions are the sub-viral agents, which function as proteinaceous infectious particles without a genomic RNA or DNA. They are the mysterious pathogens whose accumulation within neurons cause severe fatal and transmissible neurodegenerative diseases in humans and animals.
Prions solely possess PrP proteins. The term prion was coined by a scientist named Stanley Prusiner. Prion diseases are very infrequent but always fatal. Prion cause abnormal folding of the normal cells in the brain.
The symptoms of prion infection progress rapidly, which requires proper medical care to control the dissemination of infection. This post describes the meaning, structure and replication cycle of prions. You will also get to know the diseases associated with prions with diagnosis and control measures.
Content: Prions
Meaning of Prion
Prions refer to the misfolded proteins or the infectious particles, transforming the normal cells into infectious variants by some unknown mechanism. They are smaller than viruses (only observed through electron microscopy). Prions cause pores in the tissues of the brain, thereby giving it a spongy architecture. As a result, prions prevalently cause various forms (hereditary, infectious and sporadic) of neurodegenerative diseases in humans and animals.
Prions aggregate into the abnormal proteins known as amyloid fibres. The abnormal protein fibres or amyloid plaques accumulate in the infected site and ultimately cause deterioration of the cell tissues due to entanglement of neurofibrils, leading to cell death. The prion aggregates are structurally stable and resistant to many chemical and physical agents.
Structure
Prions exist in two morphologically and functionally distinct states:
PrPC
It is a cellular, non-infectious and common endogenous prion protein, which primarily affects the central nervous system and various bodily tissues. PrPC is well defined in structure. It mainly possesses an alpha-helical structure and consists of 209 amino acids (in humans) with one disulfide bond.
The normal state of prion has a molecular mass of 35–36 kDa. PrPC protein is non-sedimentable. The normal form of prion is sensitive to cellular enzymes. Proteinase K readily digests the PrPC protein.
PrP protein plays a significant function in cell adhesion and intracellular signalling in vivo. Thus, PrPC protein or the cellular protein facilitates cell-cell communication in the brain.
PrPSC
PrPC undergoes structural changes and misfolds into a PrPSC state. It possesses a high proportion of β-sheet structures instead of normal α-helices. PrPSC is the infectious state resulting in amyloid plaques formation, which ultimately leads to neurodegeneration.
Here, SC refers to scrapie (the prototypic prion disease). It is certainly polydispersive in structure. The mechanism of changing a normal prion to the misfolded or abnormal state is still unknown.
However, the abnormal state transmits its infectious properties by collapsing with the nearby protein molecules, thereby turning them into the infectious state. The misfolded structure of PrP shows resistance towards the enzymatic activity of proteases that can degrade proteins.
PrPSC protein-fibre ends function as a template, where the free protein molecules may attach, to grow the fibre. The amino acid sequence of the infectious PrPSC incorporates into the normal variants of protein in the growing fibre.
Replication Cycle of Prions
Heterodimer model: It explains how prions replicate via protein only. This hypothesis predicted that an infectious PrPSC molecule interacts with a normal PrPC molecule and mediates its transformation into PrPSC. Then, the two PrPSC molecules segregate to transform more PrPC into the abnormal PrPSC molecules.
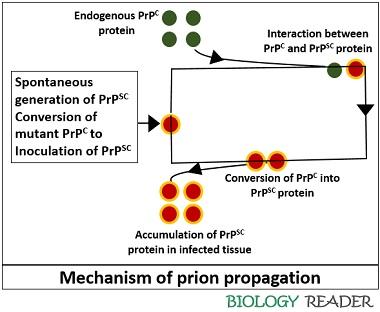
Fibril model of prion propagation: An alternative model explains that PrPSC possesses fibrillar structures whose ends bind with the normal endogenous PrPC molecules, transforming them into PrPSC.
After the conversion and propagation of prions, the aggregated infectious particles form amyloid fibres that accumulate in the infected tissue as amyloid plaques.
Therefore, it is believed that the abnormal form of prion PrPSC originates spontaneously and transmits through various ways like food ingestion and mechanical transmission. It directly interacts with an endogenous PrPC protein and enables its structural distortion.
The mechanism behind the conformational change is still not clear. However, it is believed that the cellular protein (Protein X) is associated with the transformation of PrPC to PrPSC.
The replication mechanism of prions creates implications in drug designing because of its long incubation period. An effective drug can slow down the exponential growth of prions by binding to the fibril ends and blocking them from growing further.
Prion Diseases
Prions are infectious particles, which primarily cause neurodegenerative disorders in both humans and animals.
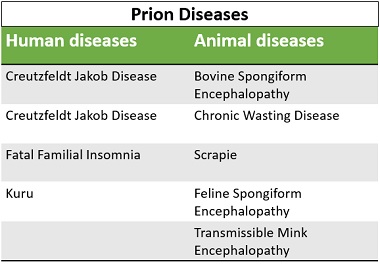
- CJD: It stands for Creutzfeldt Jakob disease. It is a kind of degenerative neurological disorder occurring in human that is invariably fatal. It leads to dementia and ultimately death. Sometimes, it also refers to the human form of “Mad Cow Disease”.
- vCJD: It stands for variant Creutzfeldt Jakob disease. It is also a neurodegenerative disease that relatively affects adults and remains for a longer duration of illness.
- Fatal Familial Insomnia: It is a heritable autosomal dominant disease of the nervous system that causing difficulty in sleeping.
- Kuru. This disease is seen in the new guinea pig that is mostly affecting women and children. In the early stage, it causes involuntary tremors, leads to dementia, and ultimately cause death.
- Bovine Spongiform Encephalopathy: It is commonly known as “Mad cow disease” that mainly affects the cow’s nervous system. It is a kind of neurodegenerative disorder that makes the brain spongy, after which a cow acts strangely and mad and lose their ability to perform involuntary functions.
- Chronic Wasting Disease: It prevalently occurs in the members of the deer family that causes chronic waste loss leading to death. The first case was seen in mule deer in wildlife research of Northern Colorado, USA.
- Scrapie: It is a neurodegenerative disease that primarily affects the nervous system of sheep and goats. It is also known as “Rida”.
- Feline Spongiform Encephalopathy: It affects the cat family members, and the disease is invariably fatal.
Risk Factors
Risk factors for prion disease include:
- Any member in the family history infected with prion disease.
- People who have consumed meat infected by prions.
- People infected via the insertion of contaminated medical equipment.
Symptoms
- Rapidly developing dementia
- Difficulty in walking
- Hallucinations
- Muscle stiffness
- Confusion
- Fatigue
- Difficulty in speaking
Diagnosis
The diagnosis of prion disease includes:
- Sample collection: Collect the sample of brain tissue during a biopsy or after death. You can also take a sample of a spinal tap or lumbar puncture and perform blood tests.
- MRI (magnetic resonance imaging) diagnose injuries in the nervous system by simply scanning the brain.
- An electroencephalogram analyzes brain waves, which requires placing electrodes on the scalp. Neurologic and visual exams check nerve damage and vision loss.
- Treatment: Prion diseases are incurable, but the administration of certain drugs can slow down the progression of the disease.
Control
The products must be labelled that contain rendered cattle or sheep. Manufacturers must prevent ruminant products from being mixed with the rendered products. You should take proper precautions by maintaining proper sanitation. If anyone is dealing with any neurological disorder, that person should not donate his organs or tissue.